
일상적인 시퀀싱 업무를 ONT로 쉽게 옮기는 방법
Easy transfer of routine, day-to-day sequencing applications to the Oxford Nanopore platform
EPI2ME 워크플로로 Amplicon, Plasmid, AAV QC, Bacterial WGS까지 한 번에
기존에 캡릴러(=Sanger)나 short-read 기반으로 “일상적으로 하던” 시퀀싱 업무들을, Oxford Nanopore 플랫폼 + EPI2ME end-to-end 워크플로로 쉽게 전환할 수 있다는 점입니다.
포스터는 크게 4가지 루틴 워크플로를 예시로 들고 있습니다.
- Amplicon 분석(amplicon-wf)
- Plasmid construct 검증(wf-clone-validation)
- AAV 유전체 QC(wf-AAV-QC)
- 세균 isolate WGS(wf-bacterial-genomes)
각 워크플로가 “샘플 준비 → 시퀀싱 → EPI2ME 분석”까지 이어지는 형태로 소개되어, 현장에서 바로 적용 가능한 “전환 가이드” 성격이 강합니다.
1) Amplicon-wf: Sanger 애매한 염기(ambiguous base)를 나노포어로 해결
포스터는 PCR amplicon 시퀀싱이 분자생물학에서 가장 흔한 루틴 작업 중 하나라고 전제합니다. 그리고 barcoding(rapid 또는 native)을 통해 스케일업이 가능하다고 설명합니다.
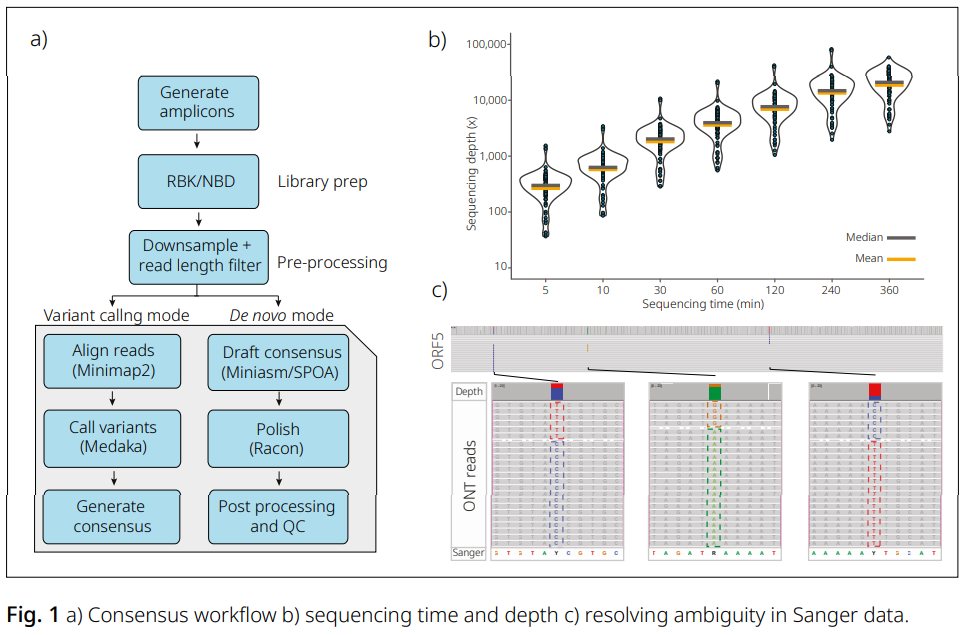
EPI2ME™ wf-amplicon을 활용한 확장 가능한 앰플리콘 분석
PCR을 이용해 유전체의 특정 영역을 증폭하는 것은 다양한 분자생물학 응용 분야에서 널리 사용되는 일반적인 절차입니다. Rapid 또는 native 방식의 바코딩을 활용하면 Oxford Nanopore 플랫폼에서 확장 가능한 시퀀싱이 가능합니다.
EPI2ME wf-amplicon(Fig. 1a)을 사용하여 돼지 생식기·호흡기 증후군 바이러스(PRRSV)의 ORF5 영역을 타겟으로 한 앰플리콘의 de novo 합의서열(consensus sequence)을 생성했습니다.
다운샘플링 결과, 단 2시간의 시퀀싱만으로도 94개 앰플리콘에 대해 1,000배 커버리지를 확보하기에 충분함을 확인했습니다(Fig. 1b).
또한 Sanger 합의서열에서 염기가 모호하게(ambiguous) 나타난 샘플을 추가 분석한 결과, 나노포어 리드를 통해 서로 구분 가능한 SNV를 가진 세 개의 집단이 존재함을 확인할 수 있었습니다(Fig. 1c).
2) wf-clone-validation: 플라스미드를 “통째로” 검증한다 (Sanger가 어려운 지점을 정면으로)
두 번째 블록은 plasmid construct 검증입니다. 포스터는 AAV transgene control plasmid(GFP-pAAV)를 예시로 들며, Rapid Barcoding으로 준비하고 EPI2ME clone-validation 워크플로로 분석했다고 설명합니다.

EPI2ME wf-clone-validation을 활용한 플라스미드 구성 검증
우리는 end-to-end 프로토콜을 따라 일반적인 AAV 트랜스진 대조 플라스미드(pAAV-GFP)를 시퀀싱하였으며, Rapid Barcoding Kit로 샘플을 준비하고 EPI2ME wf-clone-validation을 이용해 데이터 분석을 수행했습니다.
리드 길이 분포에서 약 5.3 kb 부근에 피크가 나타났는데, 이는 단일 절단(single-cut)된 전체 길이(full-length)의 pAAV-GFP 플라스미드를 의미합니다(Fig. 2a).
EPI2ME 워크플로를 사용하여 트랜스진과 ITR 서열의 존재를 확인하는 플라스미드 맵을 생성했으며(Fig. 2b), 제조사 레퍼런스와 비교했을 때 높은 정확도로 모든 주요 플라스미드 구성 요소를 확인할 수 있었습니다(Fig. 2c).
정렬 결과에서는 커버리지가 고르게 분포되어 있었으며, 이는 rapid barcoding transposase에 의해 원형 플라스미드가 한 번 절단되었음을 보여줍니다(Fig. 2d).
3) wf-AAV-QC: AAV QC에서 “truncation hotspot + 혼합군(population) + 오염원”을 한 번에
세 번째 블록은 AAV 유전체 품질관리(QC) 입니다. 포스터는 AAV QC에서 흔히 요구되는 항목으로 다음을 언급합니다.
- 합의서열(consensus) 생성
- truncation(절단/부분 유전체) 위치와 hotspot 확인
- ssAAV/scAAV 혼합 여부 확인
- 잠재적 오염원(Host, Helper, cap-rep plasmid carryover 등) 확인
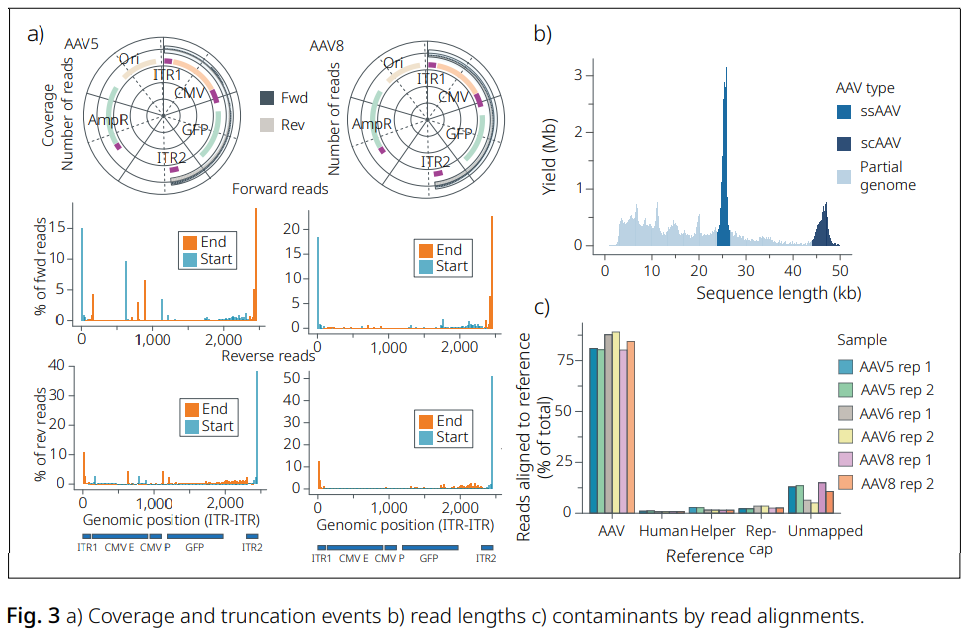
EPI2ME wf-AAV-QC를 활용한 AAV 유전체 특성 분석
AAV 제제의 품질 관리를 위해 아데노연관바이러스(AAV) 유전체의 절단(truncation) 핫스팟을 식별하고 합의서열(consensus sequence)을 생성하는 것은 일반적인 요구사항입니다.
주요 리드 정렬(primary read alignments)의 시작 및 종료 위치를 합산하여 계산하면, 절단된 유전체와 그 위치를 확인할 수 있습니다. 본 예시에서 AAV5 샘플은 CMV 프로모터 영역에서 절단이 관찰되었으며, 이러한 절단은 AAV8 샘플에서는 나타나지 않았습니다(Fig. 3a).
AAV5의 리드 길이 분포는 ssAAV와 scAAV가 혼재된 집단과 함께 잠재적인 절단 이벤트를 보여줍니다(Fig. 3b).
또한 벡터 오염의 잠재적 원인을 확인하기 위해 리드를 숙주(host), 헬퍼(helper), rep-cap 플라스미드에 정렬하여, 제조 과정 중 물질이 일부 잔존했을 가능성(carryover)을 확인하였습니다(Fig. 3c).
4) wf-bacterial-genomes: 나노포어만으로도 isolate WGS를 “정확하고 완전하게”
네 번째 블록은 세균 isolate WGS입니다. 포스터는 여기서 “나노포어만으로 만든 assembly”를 Medaka v2.0.1로 벤치마크했고, 이전 버전보다 정확도가 개선되었다고 설명합니다.
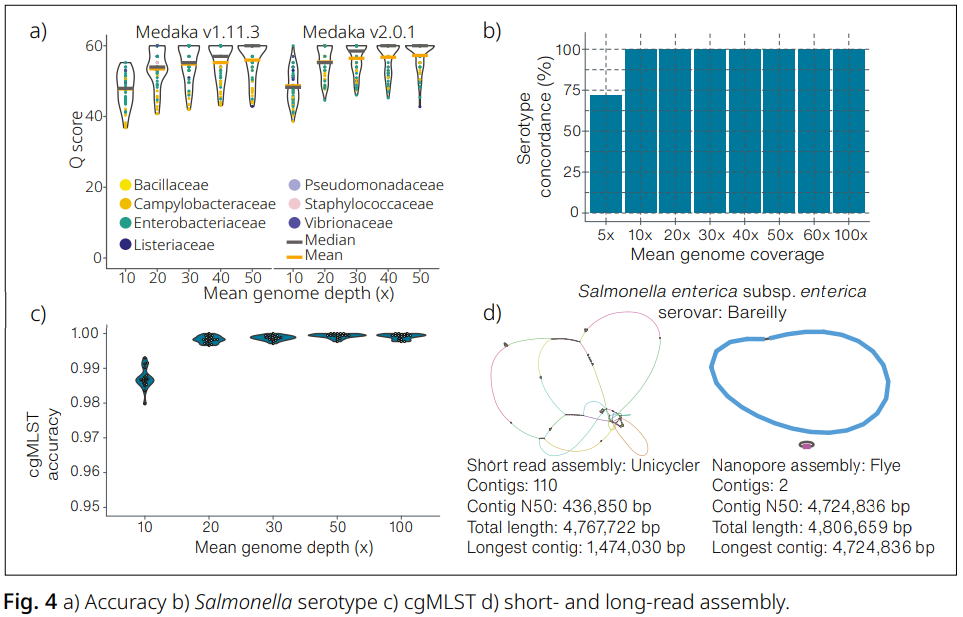
EPI2ME wf-bacterial-genomes를 활용한 세균 분리주 분석
Nanopore 단독으로 수행한 세균 분리주의 유전체 조립(genome assembly)은 Medaka v2.0.1을 사용하여, 나노포어-쇼트리드 하이브리드 조립(n=61)과 비교 평가되었습니다.
Medaka v2.0.1로 생성된 Nanopore 단독 조립은 이전 버전 대비 개선된 정확도를 보였으며, 30배 커버리지에서 Q56/Q58.5, 50배 커버리지에서 Q56/Q60 수준을 달성했습니다(Fig. 4a).
서로 밀접하게 관련된 다양한 분류군을 포함한 32개 분리주에 대해 Salmonella 혈청형 분석을 수행한 결과, 10배 커버리지에서도 기존에 알려진 혈청형과 완전히 일치하는 결과를 보였습니다(Fig. 4b).
또한 core genome MLST 프로파일은 20배 커버리지에서 99.5% 이상의 일치율을 나타냈습니다(Fig. 4c).
동일한 분리주에 대해 쇼트리드 기반 조립과 Nanopore 단독 조립을 비교한 결과, Nanopore 플랫폼만으로도 완전한 원형(contiguous circular) 컨티그가 생성됨을 확인했습니다(Fig. 4d).
https://nanoporetech.com/resource-centre/easy-transfer-of-routine-day-to-day-sequencing-applications-to-the-oxford-nanopore-platform
'나노포어 최신 동향 > 연구 · 응용 사례' 카테고리의 다른 글
| 글로벌 유전체 연구의 현재와 미래 (0) | 2026.04.16 |
|---|---|
| 왜 Nanopore가 AMR 연구의 게임체인저인가? (0) | 2026.03.17 |
| 식중독 병원체 감시의 새로운 표준:Oxford Nanopore 기반 WGS로 글로벌 조화를 이루다 (0) | 2026.02.25 |
| 2025년, Nanopore-only 미생물 WGS 정확도의 전환점 (0) | 2026.02.24 |
| Nanopore 표준 리드만으로 T2T (Telomere-to-Telomere) 인간 유전체 어셈블리 구현하기 – hifiasm 기반 새로운 접근 (0) | 2026.02.24 |